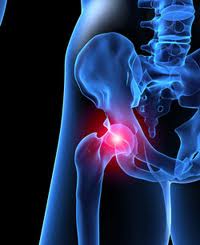



The DePuy metal-on-metal hip replacements have affected many citizens and often left them with serious and life-threatening complications. These implants suffer an extremely high failure rate, which is projected to be around 80% at just 8 years after surgery. The complications of a metal-on-metal hip replacement can be extremely severe. Problems after surgery range from pain, swelling, limited mobility, trouble walking, and dislocation, to a very serious disease called metallosis. The grinding that occurs between parts of the hip replacement can release metal debris into the body and bloodstream, causing the blood levels of chromium and cobalt to become dangerously high. This metal poisoning in the blood can cause cardiovascular, neurological, thyroid and renal issues in the body as well as destroying muscle, tissue, and bone. There have already been thousands of lawsuits filed citing the disastrous results of a DePuy metal-on-metal hip implant.
DePuy, a Johnson & Johnson company, issued a recall on these products in August of 2010. Due to Texas’ 2 year statute of limitations law, clients will no longer be able to file lawsuits against DePuy metal-on-metal hip replacements after August 23, 2012. A statue of limitations law is intended to put a specific length of time on certain lawsuits, so that future filings cannot be extended indefinitely. While this can help avoid clogging in the justice system, it may also leave many victims incapable of receiving compensation for their suffering.
If you or a loved one has been adversely affected by a DePuy metal-on-metal hip replacement, don’t let the sun set on your filing rights. Every patient should be entitled to proper care and certainly proper medical device implants. If you fail to file a lawsuit by the August 23 deadline, your claim could be lost forever. No one should have to live with life-altering complications from a defective medical device without retribution, so make sure you are one of the ones to have your voice heard. Contact Borchardt Law Firm now for a consultation about your case.
—–